Established 2005
research community




MECEA
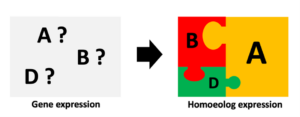
Solving the living puzzle: Homoeolog interaction in polyploid genomes
Many economically important crops, such as wheat, cotton and coffee are allopolyploids, i.e., they originated from hybridisation of two or more species followed/preceded by whole

MECEA
My Journey in Wheat
Ever since I was a teenager, I have been inspired by the “Golden Rice” story and amazed by how the application of plant sciences can
MECEA
Unravelling the genetic relationships between cereals and their microbiome
Cereals, like other plants, live in association with a myriad of microorganisms collectively referred to as the plant microbiome. These interactions are particularly dynamic in